摘 要:苎麻是多年生作物,连作多年会导致苎麻败蔸、产量下降,甚至死亡等,严重影响苎麻产业的发展。试验以连作和非连作苎麻根际土壤为研究对象,采用Illumina MiSeq测序平台,对苎麻根际土壤细菌和真菌群落结构进行分析。结果表明,细菌群落结构中厚壁菌门、变形菌门、酸酐菌门、放线菌门和拟杆菌门为苎麻根际土壤细菌的优势菌门,连作苎麻根际土壤主要细菌为厚壁菌门,占细菌总数的93.46%,而非连作苎麻根际土壤主要为厚壁菌门、变形菌门、酸杆菌门,分别占细菌总数的64.60%、13.70%、5.77%;真菌中子囊菌门和担子菌门为苎麻根际土壤的优势菌门,连作与非连作苎麻根际真菌的门类未发生改变,但是各门类的比率发生了改变;主成分分析表明,连作与非连作苎麻根际土壤微生物丰富度有明显不同。综上所述,与非连作苎麻比较,连作苎麻根际细菌和真菌群落结构以及多样性发生了改变,多样性呈现下降的趋势,且真菌群落的含量比率也发生较大的改变,因此,苎麻根际微生物群落结构和多样性的改变可能是导致苎麻连作障碍的主要因子之一。
苎麻(Boehmeria nivea L.Gaud.)属于荨麻科苎麻属多年生草本植物,是中国特有的以纤维为主要用途的经济作物。中国是苎麻的主要产区,全世界90%以上的苎麻产自中国,在国际上被称为“中国草”,印度和其他东南亚国家也有苎麻种植[1]。另外,苎麻叶片蛋白质含量高,生物产量高,是一种理想的饲料作物[2]。前期研究[3]表明,无论是作为饲料用途还是纤维用途,连作障碍都会给苎麻带来严重的危害,影响麻农种麻积极性,进而影响苎麻产业的发展。研究者前期开展了苎麻连作障碍机理的研究,初步探明苎麻连作障碍发生时期[3],明确了连作障碍主要影响因子为根腐线虫危害、根系自毒物及根际微生物等[4],并对连作障碍与健康苎麻的根际土壤开展了宏基因组和代谢组的研究,获得了影响苎麻连作障碍主要微生物种类及代谢产物[5]。但是关于连作与非连作苎麻根际微生物差异的研究较少,不同苎麻种植区域根际微生物也存在较大的差异,江西是苎麻种植的主产区,该地区连作苎麻根际微生物相关研究尚未开展。
前人研究[6,7]表明,土壤微生物数量和结构的变化直接或间接地影响土壤中养分的转化和利用。作物连作多年,土壤微生物活性下降,微生物的种群结构发生改变,有害微生物逐渐增加,养分消耗单一,直接影响作物对养分的吸收利用,养分的利用率降低[8,9]。传统的土壤微生物研究方法有生物平板培养法、Biolog 鉴定系统法、生物标记等[10],这些传统的方法往往不能全面地估计土壤微生物的群落组成,无法详细地描述出不同群体的生理差异。随着测序技术的快速发展,第二代测序Illumina MiSeq方法有效地克服了传统方法通量低、操作复杂和准确率低等缺陷[11],具有操作简单、成本较低、结果可信度高等特点。目前此平台已在微生物多样性群落结构研究方面得到了广泛应用[12,13]。近年来一些研究表明,连作导致土壤微生物结构和种群发生改变[14],Zhao等[15]报道了香子兰单一种植年限的增加会导致根际有益微生物数量减少,有害病原真菌的数量增加。Sun等[16]研究表明,连作多年的苹果树土壤根际细菌种群结构发生了很大的变化,有害微生物增加,病菌滋生比较多。由此可见,土壤微生物对连作作物具有较大的影响,健康稳定的微生物结构是作物长期连作稳定高产的基础。江西是中国苎麻主产区,但连作苎麻根际微生物群落结构特征尚不清楚,不利于构建合理的农作调控技术。为了更加明确江西地区苎麻根际微生物多样性及其种群变化,本研究以江西苎麻主产区不同连作年限苎麻根际土壤为材料,采用高通量测序技术,探讨连作与非连作苎麻地根际土壤细菌、真菌多样性的变化,以期为苎麻连作障碍调控提供理论依据。
1材料与方法
1.1研究区概况
试验地位于江西省宜春市农科院苎麻资源圃(北纬27°46′58″,东经114°24′44″),供试土壤为红壤土。选择连作10年赣苎三号苎麻土壤为试验材料(连续10年一直种植该苎麻品种),1年新栽的赣苎三号苎麻土壤为对照(对照地块之前为撂荒地,未种植任何作物,当年种植赣苎三号扦插苗)。对两块地的土壤营养氮、磷、钾的含量进行了检测,连作10年的土壤样品总氮平均含量为1.105%,全磷含量0.073%,全钾含量为3.418%,1年新栽的总氮含量为1.082%,全磷含量0.081%,全钾含量为3.415%,两块地土壤营养水平差异较小,后期两块地栽种同一品种,栽培措施、肥水管理等保持一致。
1.2试验设计
试验布设于2016年5月,选择连作10年且产生了连作障碍的赣苎三号苎麻地和1年新栽苎麻地,将整个苎麻蔸挖出来,轻轻抖落根系,去掉表土(0~5cm),再用清洁的毛刷轻轻刷取附着在根表面的土壤,此为根际土,根旁边的土壤为非根际土壤。1年新栽麻根际土壤命名为GNR,1年新栽麻非根际土壤命名为GNN,连作10年苎麻根际土壤命名为GRR,连作10年苎麻非根际土壤命名为GRN。
以新栽的1年苎麻地土壤(非连作)为对照,对连作10年的苎麻地土壤(连作)进行分析,每个处理用5点取样法取连作10年地的根际土壤混合为一个处理(GRR),连作10年非根际土混合为一个处理(GRN);新栽1年的同上取样方式,根际土壤混合为一个处理(GNR),非根际土壤混合为一个处理(GNN),每个处理3次重复,共12个样品,置于-70℃冰箱保存备用。
1.3试验方法
1.3.1土壤微生物基因组DNA的提取
提取试剂盒为Omega土壤微生物DNA提取试剂盒,称取在-20℃保存的土壤样品0.5g,按试剂盒的试验步骤进行土壤微生物总DNA的提取,DNA的质量和数量采用NanoDrop分光光度计和琼脂糖进行检测,提取的DNA稀释到1ng/μL于-20℃保存待用。
1.3.2PCR扩增及高通量测序分析
提取样品总DNA后,根据细菌V3-V4区的通用引物338F(5′-ACTCCTACGGGAGGCAGCAG-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)[17],真菌V1-V2区通用引物fITS7[18]和ITS4[19]进行PCR扩增。然后对其扩增产物进行纯化、定量和均一化形成测序文库,建好的文库先进行文库质检,质检合格的文库采用Illumina Mic-Seq测序平台(Illumina,San Diego,CA,USA)进行测序,测序由上海欧易生物科技有限公司完成。
1.4生物信息学分析
由IlluminaMiseq测序所得数据称为raw reads或raw data,用Trimmomatic软件对原始双端测序数据进行去杂并拼接,接着用FLASH软件对拼接后的paired end数据进行精准去杂[20],用QIIME软件进行序列分析得到较优质的序列[21],进行下游分析。优质序列使用CD-HIT[22]方法以97%相似度进行OTU分类,随后以OTU分类中丰度最大的序列为代表序列。与Greengenes数据库[23]比对后最终得到系统发育树和OTU分类。
1.5数据分析
随机选取相似度在97%条件下的OUT生成稀释曲线,并利用软件Mothur计算丰富度指数Chao[24],多样性指数Simpson和Shannon[25]。基于RDP和UNITE分类学数据库对OUT进行物种注释,并采用Excel和SPSS进行数据处理。
2结果与分析
2.1土壤样品测序深度评估
12个苎麻根际土壤样品测序后,细菌样品共获得的总序列数为1307718条,双端Reads拼接后共产生469848条Raw Tags,序列优化后共得到439 812条Clean Tags,经过测序数据预处理后,根据序列的相似性,将clean序列进行OUT分类共获得55001个OTUs;12个样品真菌测序,过滤后共获得1013741条序列,双端Reads拼接后共产生345788条Raw Tags,序列优化后共得到336765条Clean Tags,相似性分类共获得4321个OTUs。参照文献[26]随机抽取测序序列,将抽到的序列数与其所能代表OUT的数目构建曲线,在97%相似性水平下聚类OTU并制作各样品的稀释曲线图,细菌和真菌曲线逐渐趋向平坦,说明测序数量合理。
2.2连作、非连作苎麻根际土壤细菌和真菌群落丰富度和多样性变化
Observed species指数和Chao1指数可反映群落物种丰富度。由表1可知,种植1年的苎麻根际土壤细菌Observed species指数和Chao1指数平均分别为1806、2668.06,而连作10年的苎麻根际土壤细菌Observed species、Chao1指数平均数分别为1325、2029.69,连作10年的苎麻根际细菌指数均比1年新栽苎麻的要低,根际土壤比非根际土壤细菌Observed species、Chao1指数低;土壤真菌的这两个指数也呈现同样的变化规律,这说明苎麻根际土壤细菌和真菌群落物种丰富度变化因连作年限的不同而有较大的差异,连作多年的土壤细菌和真菌群落物种丰富度会减少。
Simpson指数和Shannon指数可反映群落物种多样性。由表1可知,1年新栽麻和连作10年苎麻根际土壤(GNR、GRR)Simpson、Shannon指数均低于非根际土壤(GNN、GRN),连作多年苎麻(GRR、GRN)土壤细菌Simpson、Shannon指数相对比1年新栽麻(GNR、GNN)有所下降;而根际真菌的这些指数变化差异不明显,这说明连作多年可能会减少土壤细菌群落物种的多样性。
表1 连作和非连作苎麻根际土壤细菌、真菌群落丰富度及多样性指数
|
微生物群落 |
样品 |
Reads |
OTU |
Observed |
Chao1 |
Simpson |
Shannon |
|
|
Raw |
Clean |
数量 |
species |
指数 |
指数 |
指数 |
||
|
细菌 |
GNR |
142316 |
132873 |
15677 |
1399 |
2346.51 |
0.86 |
5.046 |
|
GNN |
116398 |
107791 |
20306 |
2214 |
2989.61 |
0.98 |
8.673 |
|
|
平均 |
129357 |
120332 |
17992 |
1806 |
2668.06 |
0.92 |
6.860 |
|
|
GRR |
150235 |
141508 |
7203 |
546 |
871.89 |
0.81 |
3.660 |
|
|
GRN |
60899 |
57640 |
11815 |
2103 |
3187.48 |
0.96 |
7.980 |
|
|
平均 |
105567 |
99574 |
9509 |
1325 |
2029.69 |
0.89 |
5.820 |
|
|
真菌 |
GNR |
135176 |
131930 |
1169 |
407 |
465.33 |
0.96 |
6.230 |
|
GNN |
73031 |
72613 |
1224 |
436 |
494.78 |
0.96 |
6.170 |
|
|
平均 |
104104 |
102272 |
1197 |
422 |
480.06 |
0.96 |
6.200 |
|
|
GRR |
72695 |
68359 |
570 |
82 |
112.49 |
0.90 |
4.190 |
|
|
GRN |
64886 |
63863 |
1358 |
390 |
432.76 |
0.95 |
6.050 |
|
|
平均 |
68791 |
66111 |
964 |
236 |
272.63 |
0.93 |
5.120 |
|
2.3连作与非连作苎麻根际土壤微生物群落门类组成
从门分类水平看,连作与非连作苎麻根际土壤样本中检测出主要细菌门有11个,分别是厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、绿弯菌门(Chloroflexi)、芽单胞菌门(Gemmatimonadetes)、疣微菌门(Verrucomicrobia)、WS3、浮霉菌门(Planctomycetes)、硝化螺旋菌门(Nitrospirae)。其中厚壁菌门、变形菌门、酸酐菌门、放线菌门和拟杆菌门为苎麻根际土壤的优势菌门(图1),这些菌门在连作苎麻、非连作苎麻根际土壤中分别占细菌总数的84.53%、91.52%。连作苎麻根际土壤(GRR)主要菌门为厚壁菌门(Firmicutes),占细菌总数的93.46%,其次为变形菌门(Proteobacteria);而新栽1年苎麻根际土壤(GNR)主要菌门为厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria),分别占细菌总数的64.60%、13.70%、5.77%。连作与非连作苎麻非根际苎麻土壤细菌的门类和含量差异不明显,这说明连作多年苎麻根际细菌群落门类比非连作的发生了较大的变化。
测序结果表明,从苎麻根际土壤中鉴定得到的真菌主要来自6个门,包括子囊菌门(Ascomycota)、担子菌门(Basidiomycota)、接合菌门(Zygomycota)、球囊菌门(Glomeromycota)和壶菌门(Chytridiomycota)等。其中优势菌群为子囊菌门(Ascomycota)和担子菌门(Basidiomycota),在连作10年苎麻非根际(GRN)和根际土壤(GRR)中,子囊菌门占总真菌群落的43.60%、58.90%,担子菌门占总真菌群落的29.80%、30.50%,而新栽的1年苎麻地土壤非根际土壤(GNN)和根际土壤(GNR)中,子囊菌门占总真菌群落的37.40%、55.31%,担子菌门占总真菌群落的40.00%、24.39%(表2)。在连作与非连作苎麻根际土壤中,各优势菌群比率有差异,子囊菌门在连作苎麻根际比率比较大,而担子菌门差异不明显。以上结果显示,长期连作苎麻根际土壤细菌和真菌群落门类会发生改变。

图1 连作与非连作苎麻根际土壤细菌、真菌群落结构图(在门水平上)
注:A:细菌;B:真菌。
表2连作和非连作苎麻根际土壤真菌门水平分布比例
|
土壤样品 |
子囊菌门(Ascomycota) |
担子菌门(Basidiomycota) |
接合菌门(Zygomycota) |
球囊菌(Glomeromycota) |
壶菌门(Chytridiomycota) |
|
GNN |
37.40 |
40.00 |
6.28 |
1.76 |
0.29 |
|
GNR |
55.31 |
24.39 |
3.12 |
1.81 |
0.97 |
|
GRN |
43.60 |
29.80 |
7.50 |
1.79 |
0.14 |
|
GRR |
58.90 |
30.50 |
2.80 |
0.56 |
0.00 |
应用Bray-Curtis距离算法和Complete聚类法将3个系统中属水平上主要菌群中的高丰度和低丰度的物种分块聚集,通过颜色梯度及相似程度来反映多个样本在各分类水平上群落组成的相似性和差异性。如图2A所示,蓝色相似度最差,红色相似度最高,说明在连作与非连作苎麻根际土壤中细菌类群的Bacillus、Enterococcus、Lactococus这三属相似性比较大,没有显著差异,在连作与非连作土壤中Rhodoplanes、Kaistobacter、Flavobacterium、Nitrospira、DA101、Steroidobacter、Pillimelia、Methylibium等细菌属差异性比较大。从图2B可以看出连作非连作苎麻根际真菌群落的相似性,其中Candida、Cryptococcus、Mortierella及Guehomyces等相似性比较大,而Cylindrocarpon、Phoma、Staphylotrichum、Ceriosporopsis等在连作10年、新栽1年苎麻根际土壤中差异比较大,这些真菌属类可能与苎麻连作障碍存在一定的关系。
2.4连作与非连作苎麻根际土壤微生物组成及主成分分析
从连作与非连作苎麻根际微生物维恩图可以看出,在97%的相似性下,得到了每个样品的OTU个数。Venn图能够反映组间或样品之间共有和特有OTU的数目,直观地表现出组间或样品间OTU的重叠情况[26]。结合OTU所代表的物种,可以找出不同环境中的核心微生物。从图3A中可以看出,不同处理之间共有的细菌OTU数目为156个,代表的物种分别属于厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)和拟杆菌门(Bacteroidetes)等,各处理间差异比较大;从图3B中可以看出,不同处理之间共有真菌OUT数目为43个,代表的物种分别属于接合菌门(Zygomycota)、担子菌门(Basidiomycota)、子囊菌门(Ascomycota)等,不同的处理间真菌的门类也存在较大的差异。
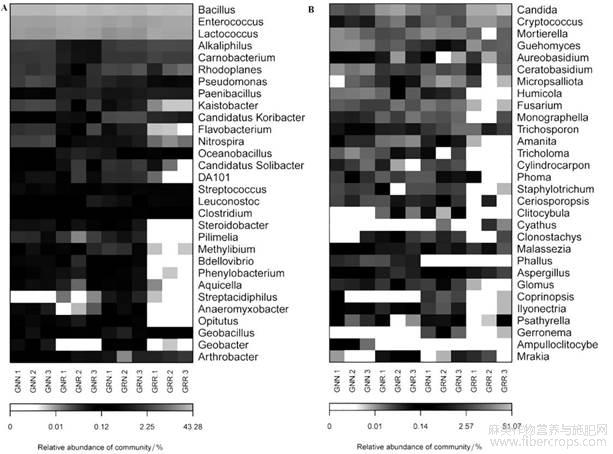
图2连作与非连作苎麻根际土壤属水平微生物群落热点图

图3连作与非连作苎麻根际微生物群落OTUs维恩图
从图4A可以看出,各种颜色代表了连作、非连作苎麻根际土壤3个重复的样品,其中两个样品距离越近表明这两个样品物种组成越相似,从细菌门类的物种组成来看,非连作苎麻根际细菌(GNN,GNR)组成比较类似,而连作10年苎麻非根际(GRN)的物种组成与连作根际(GRR)的距离较远,表明他们之间的物种组成差异性较大;从图4B可以看出,连作10年(GRR、GRN)与1年新栽麻(GNN、GNR)苎麻根际真菌的组成具有一定的差异,但差异不显著。

图4连作与非连作苎麻根际微生物主成分分析图
注:A:细菌;B:真菌。
3讨论与结论
连作障碍也叫再植病或土壤病,是指同一地块连续多年种植相同作物造成作物生长状况变差、产量和品质降低、病虫害发生加剧的现象。研究[27]表明,造成作物连作障碍的因素是多方面的,比如土壤理化性质的改变、土壤营养物质的失衡及自毒物质的积累等,但也有研究者认为,根系分泌物生态效应的间接作用及土壤微生物区系的变化是导致植物连作障碍形成的主要因素[28]。这可能是由于在根系分泌物特定组分的介导下,某些类群的微生物(如土传病原菌)大量繁殖,同时抑制其他有益微生物(如假单胞菌等拮抗菌)的生长,进而改变了植物根系分泌物的组分和数量,为趋化性病原微生物提供更多的碳源、能源,形成恶性循环,造成植物生长发育不良。
苎麻是多年生作物,连作多年会产生苎麻连作障碍,通过前期的研究明确了根腐线虫害是导致苎麻连作障碍的主要因子,通过对根腐线虫害苎麻全基因组表达谱进行研究,获得了受根腐线虫害影响的基因[29]。但是,关于连作障碍对苎麻根际微生物区系的影响研究较少,本文采用Illumina MiSeq 测序平台,对连作、非连作苎麻根际土壤细菌和真菌群落结构进行分析。结果表明,厚壁菌门、变形菌门、酸酐菌门、放线菌门和拟杆菌门为苎麻根际土壤细菌的优势菌门,连作苎麻根际土壤主要菌门为厚壁菌门,占细菌总数的93.46%,而非连作苎麻根际土壤主要菌门为厚壁菌门、变形菌门、酸杆菌门,分别占细菌总数的64.60%、13.70%、5.77%。由此可见,连作与非连作苎麻根际细菌门类和比例都发生了较大的变化,连作10年苎麻根际土壤大部分为厚壁菌门,厚壁菌门被分为三个纲:厌氧的梭菌纲、兼性或者专性好氧的芽孢杆菌纲。冯伟等[30]研究表明,芽孢杆菌属是天津团泊湖可培养耐盐碱菌的优势细菌种群。已有研究[31]报道芽孢杆菌可以产生内生孢子,在干燥、热、紫外辐射等不利环境下维持自身对外界的抵抗能力,变形菌门和厚壁菌门是许多盐渍化土壤中的优势菌群[32]。连作10年以上苎麻根际以厚壁菌门、变形菌门为主,可能是由于在不利的根际环境下产生大量的芽孢杆菌来维持自身不受伤害。此研究与吴林坤等[33]研究相似,不同连作年限地黄根际土壤细菌群落结构存在一定差异,野生状态地黄土壤和头茬土壤菌群较为相似,变形菌门和厚壁菌门占据优势地位。
真菌中子囊菌门和担子菌门为苎麻根际土壤的优势菌门,连作与非连作苎麻根际真菌的门类没有发生改变,但是各门类的比率发生了改变,因此真菌各门类比率的变化是影响苎麻连作障碍的因子之一。维恩图分析也进一步验证了苎麻根际细菌和真菌门类的变化特点。赵帆等[34]研究也获得了相似的研究结果,随连作年限的延长,草莓根际土壤生态系统中细菌和真菌群落各门类组成的比例会发生显著变化。
从属的角度分析,在连作与非连作土壤细菌中Rhodoplanes、Kaistobacter、黄杆菌属(Flavobacterium)、硝化螺旋菌属(Nitrospira)、DA101、Steroidobacter、Pillimelia、Methylibium等属和真菌中柱孢属(Cylindrocarpon)、茎点霉属(Phoma)、大孢圆孢霉属(Staphylotrichum)、Ceriosporopsis属差异性比较大。黄杆菌属为抑制土传青枯病发生的根际土壤中的优势菌属,与抑病能力密切相关[35]。吴凡等[36]研究表明硝化细菌能够有效促进土壤中碘普罗胺降解,这些具有降解作用的细菌相对丰度增加,说明土壤微生物发生了变化。苎麻连作10年与非连作土壤微生物中黄杆菌属、硝化螺旋菌属等差异较明显可能会影响细菌的相对丰度和结构变化。
高通量测序序列分析中有部分未被描述和鉴定的细菌和真菌,即在与现有数据库中已知序列进行比对时,无法获得该序列的分类学信息。因此,推断这些序列可能是苎麻根际土壤中新发现的土壤微生物类群,还需采用其他分析手段进一步鉴定。
参考文献
[1]LIU T M,ZHU S Y,TANG Q M,et al.De novo assembly and characterization of transcriptome using Illumina paired-end se-quencing and identification of CesA gene in ramie (Boehmeria nivea L.Gaud)[J].BioMed Central,2013,14(1):125.
[2]Machin D H.Ramie as an animal feed:a review[J].Tropical Science,1977,19(4):187-195.
[3]朱四元,刘头明,唐守伟,等.连作苎麻的部分生理生态及细胞学观察[J].湖南农业大学学报,2012,38:360-365.
[4]朱四元,刘头明,唐守伟,等.不同连作障碍因子对苎麻农艺性状的影响[J].中国麻业科学,2014,36:137-141.
[6]傅佳,李先恩,傅俊范.重茬种植西洋参对其根区土壤微生物与土壤理化性质影响[J].微生物学杂志,2009,29(2):63-66.
[7]周陈,李许滨,杨明开,等.冬小麦不同生育期土壤微生物及养分动态变化[J].西北农业学报,2008(3):113-116.
[11]Kozich J J,Westcott S L,Baxter N T,et al.Development of a dual-index sequencing strategy and curation pipeline for analyzing amplic-on sequence data on the MiSeq Illumina sequencing platform[J].Applied and Environmental Microbiology,2013,79(17):5112-5120.
[13]Konstantinidis K T,Tiedje J M.Genomic insights that advance the species definition for prokaryotes[J].Proceeding of the NationalAc-ademy of Sciences of the United States of America,2005,102(7):2567-2572.
[16]SUN J,ZHANG Q,ZHOU J,et al.Illumina amplicon sequencing of 16S rRNA tag reveals bacterial community developmentin the rhizosphere of apple nurseries at a replant disease site and a new planting site[J].The Public Library of Science One,2014,9(10):e111744.
[19]White T J,Bruns T,Lee S J W T,et al.Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics.PCR Pr-otocols:A Guide to Methods and Applications[M].Academic Press,1990:315-322.
[25]Shannon C E,Weaver W.The mathematical theory of communication.University of Illinois Press,Urbana[J/OL].M.d.computing Computers in Medical Practice,1963.https://www.researchgate.net/publication/284180912_The_Mathematical_Theory_of_Communication_University_of_Illinois_Press_Urbana.DOI:10.1002/j.1538-7305.1948.tb01338.x.
[27]张重义,林文雄.药用植物的化感自毒作用与连作障碍[J].中国生态农业学报,2009,17(1):189-196.
[28]QI J J,YAO H Y,MA X J,et al.Soil microbial community composition and diversity in the rhizosphere of a Chinese medicinal plant[J].Communications in Soil Science and Plant Analysis,2009,40(9-10):1462-1482.
[30]冯伟,孙瑞,高广海,等.天津团泊湖地区盐碱土壤中可培养微生物群落结构和归属分析[J].农业环境科学学报,2013,32(5):1028-1035.
[32]Vasim A,Verma M K,Shashank G,et al.Metagenomic profiling of soil microbes to mine salt stress tolerance genes[J].Frontiers in Microbiology,2018,9:159.
[33]吴林坤,黄伟民,王娟英,等.不同连作年限野生地黄根际土壤微生物群落多样性分析[J].作物学报,2015,41(2):308-317.
[34]赵帆,赵密珍,王钰,等.不同连作年限草莓根际细菌和真菌多样性变化[J].微生物学通报,2017,44(6):1377-1386.
[35]沈宗专,黄炎,操一凡,等.健康与罹患青枯病的番茄土壤细菌群落特征比较[J].土壤,2021,53(1):5-12.
[36]吴凡,高品,薛罡,等.硝化细菌对碘普罗胺的降解及作用机制[J].环境工程学报,2014,8(6):2225-2230
文章摘自:白雪花,陈晓蓉,陈新范,王延周,刘头明,唐守伟,朱四元.连作与非连作苎麻根际微生物的多样性研究[J].中国麻业科学,2021,43(05):222-230.
