摘 要:大麻是一年生草本植物,一种多用途、可持续的作物。迄今为止,关于大麻遗传结构的研究还很少。在此,通过EST-SSR分子标记分析大麻的遗传多样性和种群结构。结果表明,使用20对引物共扩增出113个评分条带,其中113个(100%)是多态性的;共检测到232个等位基因,平均每对引物检测到4.0176个等位基因;观测杂合度(Ho)平均为0.7102,期望杂合度(He)平均为0.6935;200个个体香农信息指数介于0.7204~2.4625之间,平均值为1.5368;多态信息含量(PIC)变化范围为0.3519~0.8801,平均为0.6558;平均基因流(Nm)平均值为13.6525。大麻基于种群遗传结构、主坐标分析和带算术平均值的未加权对组法(UPGMA)分析,将材料聚类为三组。聚类方法之间的结果相似,但三种模型的少数个体植物分布不同。聚类结果、基因多样性和遗传相似系数表明,大麻个体总体亲缘关系较为密切。同时用5对核心引物能够区分参试种质,并为每份种质构建了指纹图谱。研究结果为今后的大麻育种、遗传改良和核心种质资源收集提供了参考。
关键词:大麻;EST-SSR;遗传多样性;种群结构
大麻(Cannabis sativa L),俗称汉麻,是一种草本植物,属于大麻科。它被认为是最古老的栽培植物之一,可用于多个应用领域,从农业和植物修复到食品、饲料、化妆品、建筑和制药行业。事实上,从这种用途广泛的植物中,可以获得各种具有工业价值的产品,例如纤维和碎屑;生物建筑和隔热材料;具有重要营养和功能特性的种子、面粉、油和具有药理学意义的生物活性化合物[1]。
遗传多样性是生物多样性的重要组成部分,是生态系统和物种多样性的基础[2,3,4]。了解种质资源的遗传多样性和结构有助于高效、合理地开发、保护和利用种质资源[5,6],目前,研究人员将多种作物的遗传多样性和种群结构研究作为重要的基础研究,并且开展了许多的相关研究。经常使用形态学和农艺学特征检测遗传变异,这些特征通常表现出受环境因素强烈影响的多基因遗传。除了环境差异外,地理隔离、系统地理学模式、基因流动和种群动态也会导致选择压力,从而导致空间结构遗传变异。由于基因组中基因的适应性变化,物种将在表型和物候方面进行适应性进化[7,8]。然而,由于缺乏大量非模式物种的基因组信息,无法确定多个候选基因。这些候选基因在其基因组的局部适应中起着重要作用,因此不容忽视[9]。分子标记分析为这种方法提供了一种有效的替代方法。目前,最常见的遗传多样性分子标记包括SNP、RFLP、RAPD、ISSR、简单序列重复序列(SSR)和AFLP[10]。
使用每种技术的选择受到诸如应用的难易程度、基因组覆盖率、成本和自动化兼容性等因素的影响。简单序列重复分子标记是共显性的,符合孟德尔定律。它操作简单,具有高度的重现性和可靠性,能够揭示子代和亲本之间不受基因表达、培养条件或环境条件影响的遗传差异,并且可以显示出大量的多态性[11,12]。SSR分子标记广泛应用于植物种质鉴定、遗传多样性、遗传连锁图谱构建、基因定位和克隆以及数量性状位点分析(QTLs)[13,14,15,16]。由于表达序列标签(EST)-SSR标记来自转录区域,因此它们具有很高的成功扩增率和相关基因注释[17]。使用SSR分子标记的遗传多样性分析已广泛应用于多种作物[15,18,19,20,21],如多年生黑麦草、紫花苜蓿和小麦,信朋飞等[22]利用SSR标记对大麻种质资源进行指纹图谱的构建。大量文献表明,SSR分子标记结果可以揭示近缘物种的亲缘关系,鉴定品种[23]。
表达序列标签是剖析复杂性状以及估计分子多样性和种群结构的基础[24],由于表达序列标签(EST)-SSR标记来自转录区域,因此它们具有很高的成功扩增率和相关基因注释[17]。SSR标记价格低廉且易于通过聚合酶链反应(PCR)检测,因此作为有价值的分子标记被广泛应用于群体筛查。本研究基于前期开发的大麻EST-SSR分子标记[22],筛选适用于大麻纤维用、籽用和花叶用类型及其种群遗传多样性和亲缘关系的核心引物,并构建指纹图谱,以期对大麻品种鉴定及种质创新提供依据。
1材料与方法
1.1实验材料
实验材料为黑龙江省科学院大庆分院认定品种及搜集保存的200份大麻种质资源(附表1),包括纤维用类型78份、籽用类型63份、花叶用类型59份。
1.2DNA提取
选取大麻叶片0.5g,运用柱式植物组织基因组DNA抽提试剂盒提取DNA。-20℃保存备用。
1.3EST-SSR引物设计及PCR扩增
基于前期开发的EST-SSR引物中随机筛选出40对,由上海生工生物技术有限公司合成。选取8个品种的DNA混合样品,对SSR引物进行多态性筛选,筛选出条带清晰、稳定性好的20对引物用于所有材料的PCR扩增,引物序列见附表2。试验所用试剂均购自上海生工生物技术有限公司。
PCR反应体系为25μL,其中DNA1μL(20-50ng/μL),10×TaqBuffer(withMgCl2)2.5μL,引物(10Umol/L)各0.5μL,Taq酶0.2μL(5U/μL),dNTP(mix)0.5μL(5μmol/L),其余用ddH2O补足。PCR扩增程序为95℃5.0min;94℃30s,60℃30s,72℃30s,30个循环;72℃延伸10min。PCR扩增产物的片段大小采用QIAxcel高级毛细管电泳仪检测。
1.4数据分析
利用BioCalculator软件通过计算各峰的峰数、峰高、峰宽、峰面积等特征,对扩增产物的单次数据进行准确分析。选用的5对引物(E20、E24、E26、E31、E17)扩增出的DNA片段按从小到大的顺序排列,统计分辨率高,条带清晰,“1”表示有片段,“0”表示无片段。
取小于95%的条带频率,使用Excel2021计算多态性条带数(NPB)和多态性条带百分比(PPB)。GeneAlEx6.51b2[25]软件转换各种文件格式进行不同分析,计算遗传多样性参数,包括等位基因数(Na)、有效等位基因数(Ne)、香浓信息指数(I)、基因流(Nm)和遗传分化系数(Fst)。利用PICCalc软件计算各引物的多态性信息含量PIC值。使用GeneAlEx6.51b2软件进行遗传距离分析、主坐标分析(PCoA),基于Nei的无偏遗传距离矩阵与MEGA5.1进行未加权的算术平均对组法(UPGMA)聚类分析[26]。
使用STRUCTURE2.3.4[27]软件分析种群遗传结构,使用基于模型的聚类算法实现贝叶斯框架和马尔可夫链蒙特卡罗(MCMC)算法。为了确定最佳的亚群数量(K),对范围从2到10的每个K值进行了五次独立运行[28]。每次运行都包含10,000步的老化期,然后是100,000次MCMC迭代。根据Evanno等人开发的模型,估计ΔK参数基于连续K值之间数据对数概率的变化率,以确定最佳K[29]。
2结果
2.1简单序列重复标记的多态性
在PCR反应中,使用20对SSR引物对200个个体共扩增出113个位点,全部能表现出多态性。每个引物组合的多态位点数量从2到9不等,平均为5.65个位点(表1)。20对引物的扩增片段大小不一,在100~300bp之间变化。所有引物对均具有较高的基因多样性值,鉴定出较高水平的多态性。平均有效等位基因数(Ne)为4.0176个。每个基因座的平均有效Na为11.6。引物还表现出较高的香农信息指数(I),200份植物材料的香农信息指数介于0.7204~2.4625之间,平均值为1.5368。PIC及香农指数最高值均为E24引物。观测杂合度(Ho)的变化范围为0.3015~0.905,平均值为0.7102,期望杂合度(He)在0.3898~0.8913之间,平均值为0.6935。平均基因流(Nm)平均值为13.6525,表明存在基因交流(Nm>1)。
表1 20条大麻EST-SSR引物的多态性分析
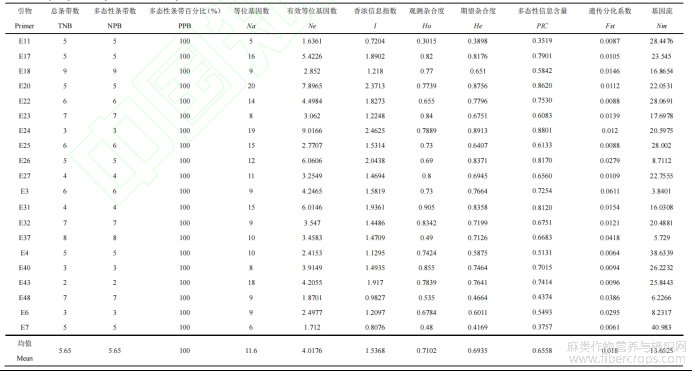
2.2不同种群遗传多样性分析
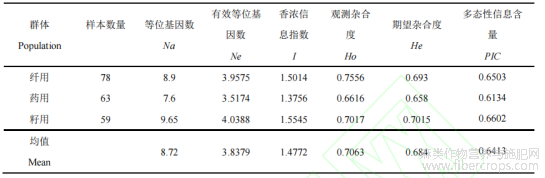
对3个参试群体进行遗传多样性指数分析,结果显示(表2),3个种群平均观测杂合度(Ho)为0.7063,期望杂合度(He)为0.684,期望杂合度均大于观测杂合度,说明各种群内均存在一定程度近交。籽用型群体的等位基因数(Na)为9.65,香浓信息指数(I)为1.5545,期望合度(He)为0.7015,观测杂合度(Ho)为0.7017,PIC值为0.6602,均高于其他两个群体,说明籽用型群体遗传多样性与其他两个类型相比较高。
表2 3个大麻种质群的遗传多样性比较分析
2.3群体间的遗传距离和遗传相似度
将200份大麻材料按用途划分为3个居群进行遗传距离比较(表3),如表所示,3个种群间的遗传距离在0.0314~0.0805范围内,Nei's遗传一致性在0.9227~0.9691范围内。籽用型种群与纤用型种群遗传距离最小,基于遗传距离构建的UPGMA聚类树也验证了籽用型与纤用型种群亲缘关系较近(图1)。同时分析三个种群的Nm和Fst(表4)观察到“花叶用”与其他人群的Nm相对较小,Fst较大,这可以解释为什么“花叶用”种群被单独归为一组。
表3 3个群体Nei's遗传一致度和遗传距离的无偏估计
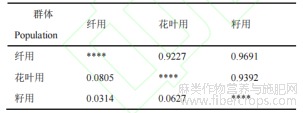
注:****上方为Nei遗传一致度,****下方为Nei遗传距离
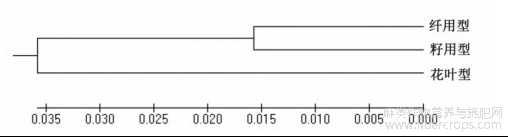
图1 基于遗传距离构建的UPGMA聚类树
表4 三个种群之间的基因流遗传分化系数

注:****上方为Nm,****下方为Fst
2.4种群结构分析
通过UPGMA聚类分析、PCoA分析和遗传结构分析,进一步探讨了基于遗传距离的不同类群和亚类种质之间的关系。使用Structure2.3.4软件分析了从200个大麻个体扫描的20个SSR引物对的标记。使用横坐标作为K值(K=1~9)和纵坐标作为lnP( K )建立折线图。lnP( K )随着K的增加而不断变化,没有最大值,无法确定最佳亚群数(图2A)。根据Evanno等[29] 人的方法确定最佳分类号。当deltaK值随K值变化时,一个明显的峰值可以确定为最佳分类数(图2B)。当K=3时,划分为3个类群(图2C)。其中绿色类群113份、红色类群44份、蓝色类群43份(图3)。
UPGMA树状图还表明,种质可以分为三个簇(图4)。200份大麻的聚类个体材料与种群遗传结构分析结果基本相符。基于UPGMA树状图,组群I主要为纤用型大麻资源,组群Ⅱ花叶型资源居多,组群Ⅲ主要为籽用型资源。
对总共200份大麻单株进行了主成分分析(图5)。他们在图中位置的距离代表亲缘关系的距离。PCoA结果与UPGMA树状图和种群结构分析结果基本一致。3组植物单株物质分布相对集中,表明它们之间的亲缘关系密切。
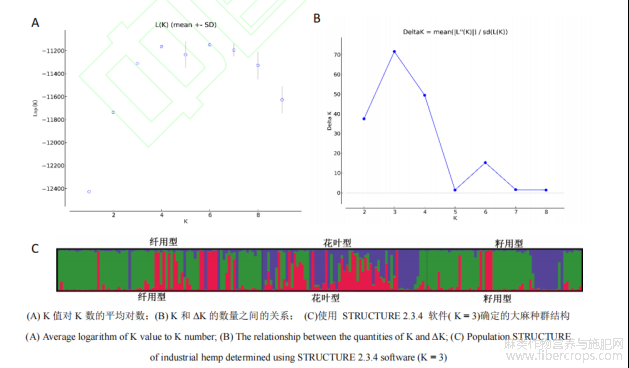
图2 200份大麻种群结构分析
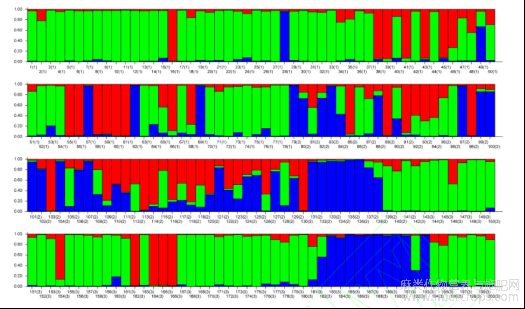
图3基于Structure的大麻核心种质资源群体遗传结构图
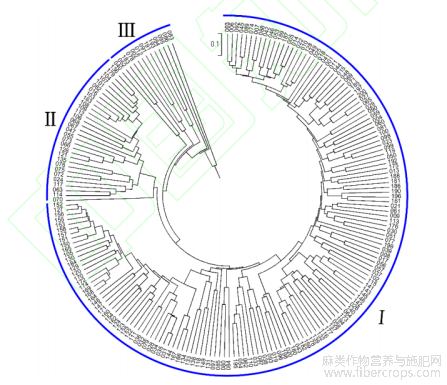
图4 基于UPGMA的200份大麻资源聚类分析
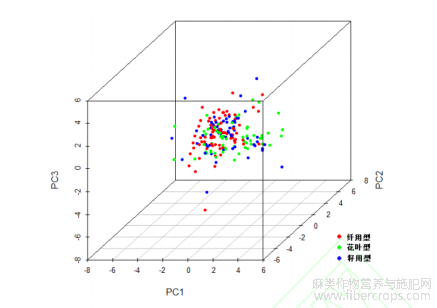
图5 200份大麻资源遗传多样性的主成分分析
2.5指纹图谱的构建
经统计分析,5对引物在200份材料中共检测到25条清晰条带。将筛选出的5对核心引物依次编号为A~E,依据5对引物扩增的多态性位点,按照指纹图谱构建方法为每一份材料建立SSR指纹图谱代码(表5)。
表5 200份大麻种质指纹图谱 导出到EXCEL


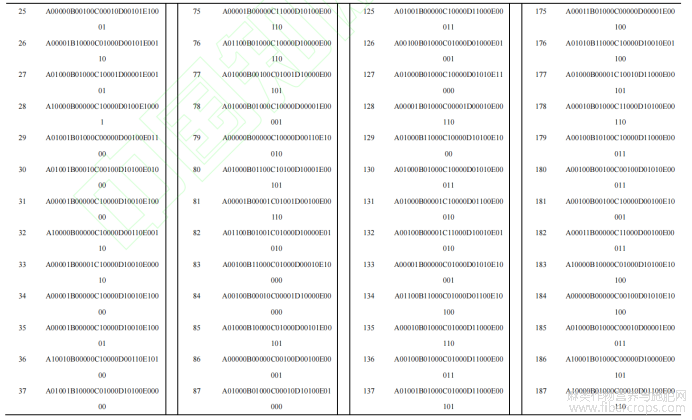
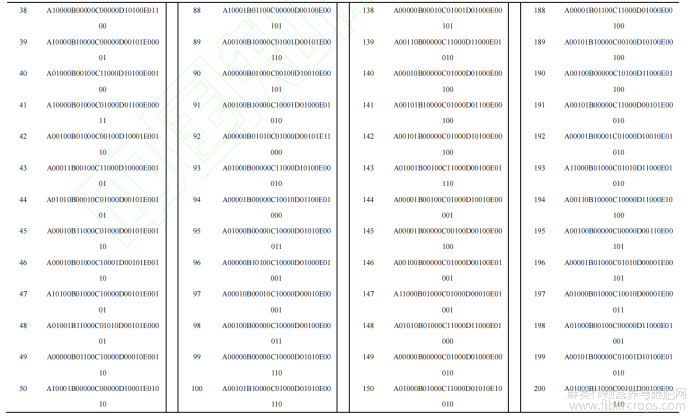
3讨论
3.1分子标记多态性分析
EST-SSR是利用已有的EST序列通过电子筛选鉴定SSR,然后进行PCR检测[30]。使用EST开发SSR,避免了开发SSR引物过程中的克隆和测序步骤,充分利用现有数据,降低了开发成本[31]。EST-SSR保守性好,在不同物种间具有良好的通用性,可以区分亲缘关系较近的材料。
探索性条件下基因型间的多态性率被认为是衡量DNA标记多样性分析效率的关键因素。大量研究指出,标记的多态性影响植物的遗传多样性水平。一般而言,使用多态性高的引物比使用多态性差的引物,供试材料的遗传参数更可靠。信朋飞等[22]。基于大麻EST信息建立SSR标记,为大麻遗传多样性研究提供理论依据。而本研究首次利EST-SSR标记对大麻种群结构进行分析,从40条EST-SSR引物中筛选出20对引物对大麻种群进行标记。本研究中检测到的高遗传多样性可能是由于使用了为大麻基因组开发的EST-SSR分子标记,可以更好地区分基因位点。香农信息指数(I)介于0.7204至2.4625之间,平均值为1.5368。这一发现表明所选引物能够客观地揭示大麻种质资源的遗传多样性。总体而言,20对EST-SSR引物可以充分分析大麻材料之间的遗传差异。
3.2大麻材料遗传变异及亚群间遗传多样性
本研究根据大麻种质资源用途类型,将其分为3个亚群进行遗传结构分析。各亚种群香农信息指数介于1.3756~1.5545之间,遗传多样性水平较高。大麻种群的高度遗传多样性可能与该物种的异花授粉有关。有研究表明PIC≥0.5,为高度多态位点;0.25<PIC<0.5,为中度多态位点;PIC≤0.25,为低度多态位点。在本研究中,20对引物PIC值平均为0.6558,说明大麻EST-SSR标记均表现为高高度多态性,说明适合大麻遗传多样性分析。
传统上,可以中和种间分化和种内遗传漂变的高度基因流动在异花授粉植物中极为常见,从而导致个体或种群之间的遗传多样性较低[17]。在我们的研究中,花叶用与其他种群之间的大麻基因流(Nm)较小,其他种群之间的Nm较高。Elam[32]提出,Nm<1表明植物种群间遗传分化程度高,而Nm>1表明植物种群间遗传分化程度低。George[33]等也在异花授粉的白三叶草中发现了这个结果。由于亚种群间基因交换频繁,种群间的亲缘关系得以维持,因此种群间的遗传分化不显著。在进化过程中,大麻发生了巨大的遗传变异,与其他种群的遗传物质频繁交换。因此,在遗传育种层面形成了多态性丰富、遗传变异度高的种质资源。不同类型的种质资源收集和引进历史也可能影响基因流动并塑造新的亚种群遗传结构。
物种的遗传结构受多种因素的相互作用影响,例如种子和花粉的传播模式、种群统计历史、地质事件、地理或生态障碍以及环境因素的发散选择[34]。根据3个群体两两之间的Nei's遗传距离(genetic distance,GD),这3个种群被分为两组。籽用型和纤用型亲缘关系最近,同时在田间测评过程中发现籽用型和纤用型品种其外部形态特征较为相似,这可能是由于在育种过程中,将来源相同的品种资源按照不同用途而划分为不同的遗传分支。
3.3大麻种质资源种群结构
本研究根据种群遗传结构分析结果,将200份材料组装成3个类群,并将来自同一种群的单株也聚类到不同的类群中。在UPGMA聚类图中,花叶用类型的一些单株与籽用型和纤用型单株聚在一起,但在种群结构分析中,花叶用材料独立聚类一组。聚类可能是育种和驯化的结果,具有对多样性结构影响很大。选择和育种倾向于使植物保持具有经济价值的性状[35]。此外,不同的环境也会引起遗传变化,从而影响种群结构的划分。也可能是大麻材料含有不同植物个体的遗传物质,所有个体的遗传信息由种群结构整合而成。UPGMA聚类是基于大麻材料的GD进行的,将密切相关的材料聚为一组,这可能导致具有不同遗传结构的材料聚集。Shen等[36]分析了64份燕麦种质资源的群体遗传结构,也发现同一种族的种质资源聚类成不同的类群。
根据PCoA、UPGMA和STRUCTURE分析,本研究中的大麻种质基本被分为三组。200份大麻单株材料的分类基本符合群体遗传结构分析,但也存在一定差异。这可能是由于不同方法应用了不同的统计原理[15]。PCoA可以根据原始数据的相异矩阵提供更有效的分类,这并不严格符合Hardy-Weinberg平衡假设。种群结构分析可以更好地了解遗传多样性,估计种质资源的变异情况,有利于对其进行有效利用。结构通过贝叶斯聚类方法按概率将种质分配给亚群,并用于自然异交种群的细分。使用UPGMA分析的种质聚类是基于遗传距离实现的,它显示了种质之间更详细的关系。总的来说,这三种方法可以共同提供对大麻的全面了解种群遗传结构。此外,所揭示的大麻差异性可以评价其在选择亲本组合时具有较高的育种和杂交优势,为选择具有强遗传差异的大麻杂交组合提供依据。
3.4SSR标记构建指纹图谱
SSRs作为一类具有等位变异高、共显性、检测简单快速、稳定性好等优点的分子标记,已在遗传多样性分析、指纹构建、性状标记和遗传连锁图谱构建等多个领域得到成熟应用[37]。许多农学家和遗传学家对SSRs进行了广泛的研究和应用。本研究选择的5对核心引物,其PIC值位于0.7901~0.8801之间,均属于高多态性引物,有利于品种资源鉴定。
4结论
本研究利用EST-SSR标记对大麻的遗传结构进行分析,并且对200份大麻种质资源进行了指纹图谱的构建。研究结果表明,籽用型种群与纤用型种群遗传距离最小,基于遗传距离构建的UPGMA聚类树也验证了籽用型与纤用型种群亲缘关系较近;同时通过UPGMA聚类分析、PCoA分析和遗传结构分析,进一步确定了200份大麻的聚类个体材料与种群遗传结构分析结果基本相符。分类结果、基因多样性和遗传相似系数表明,大麻个体总体亲缘关系较为密切。同时本研究选取5对核心引物对参试种质构建大麻指纹图谱,利用组合构成了大麻特有的DNA指纹,能够将这些材料逐一的区分开来。结果证实,大麻种质具有足够的遗传多样性。研究结果将为大麻杂交组合、标记辅助改良、种质资源保护和核心种质收集提供分子依据。
参考文献
[1]Irakli M, Tsaliki E, Kalivas A, Kleisiaris F, Sarrou E, Cook C M. Effect οf Genotype and Growing Year on the Nutritional, Phytochemical, and Antioxidant Properties of Industrial Hemp (Cannabis sativa L.) Seeds. Antioxidants (Basel, Switzerland), 2019, 8(10):
[2]Bailey J K, Schweitzer J A, Ubeda F, Koricheva J, Leroy C J, Madritch M D, Rehill B J, Bangert R K, Fischer D G, Allan G J, Whitham T G. From genes to ecosystems: a synthesis of the effects of plant genetic factors across levels of organization. Philosophical transactions of the Royal Society of London Series B, Biological sciences, 2009, 364(1523): 1607-1616.
[3]Haddad N M, Crutsinger G M, Gross K, Haarstad J, Tilman D. Plant diversity and the stability of foodwebs. Ecology letters, 2011, 14(1): 42-46.
[4]Zhang C, Vornam B, Volmer K, Prinz K, Kleemann F, Köhler L, Polle A, Finkeldey R. Genetic diversity in aspen and its relation to arthropod abundance. Frontiers in plant science, 2014, 5(806).
[5]Costa R, Pereira G, Garrido I, Tavares-De-Sousa M M, Espinosa F. Comparison of RAPD, ISSR, and AFLP Molecular Markers to Reveal and Classify Orchardgrass (Dactylis glomerata L.) Germplasm Variations. PloS one, 2016, 11(4): e0152972.
[6]Sork V L, Aitken S N, Dyer R J, Eckert A J, Legendre P, Neale D B J T G, Genomes. Putting the landscape into the genomics of trees: approaches for understanding local adaptation and population responses to changing climate. 2013, 9(4): 901-911.
[7]Feng X J, Jiang G F, Fan Z. Identification of outliers in a genomic scan for selection along environmental gradients in the bamboo locust, Ceracris kiangsu. Scientific reports, 2015, 5(13758.
[8]Rellstab C, Gugerli F, Eckert A J, Hancock A M, Holderegger R. A practical guide to environmental association analysis in landscape genomics. Molecular ecology, 2015, 24(17): 4348-4370.
[9]Li Y, Zhang X X, Mao R L, Yang J, Miao C Y, Li Z, Qiu Y X. Ten Years of Landscape Genomics: Challenges and Opportunities. Frontiers in plant science, 2017, 8(2136).
[10]Li H, Ma Y, Pei F, Zhang H, Jiang M J E J O B. Large-scale advances in SSR markers with high-throughput sequencing in Euphorbia fischeriana Steud. 2021, 49(50-55).
[11]Wang K, Lin Z, Wang L, Wang K, Shi Q, Du L, Ye X. Development of a set of PCR markers specific to Aegilops longissima chromosome arms and application in breeding a translocation line. TAG Theoretical and applied genetics Theoretische und angewandte Genetik, 2018, 131(1): 13-25.
[12]Daudi H, Shimelis H, Mathew I, Oteng-Frimpong R, Ojiewo C, Varshney R K. Genetic diversity and population structure of groundnut (Arachis hypogaea L.) accessions using phenotypic traits and SSR markers: implications for rust resistance breeding. Genetic resources and crop evolution, 2021, 68(2): 581-604.
[13]Ren R, Xu J, Zhang M, Liu G, Yao X, Zhu L, Hou Q. Identification and Molecular Mapping of a Gummy Stem Blight Resistance Gene in Wild Watermelon (Citrullus amarus) Germplasm PI 189225. Plant disease, 2020, 104(1): 16-24.
[14]Chen C, Chang J, Wang S, Lu J, Liu Y, Si H, Sun G, Ma C. Cloning, expression analysis and molecular marker development of cinnamyl alcohol dehydrogenase gene in common wheat. Protoplasma, 2021, 258(4): 881-889.
[15]Wu F, Ma S, Zhou J, Han C, Hu R, Yang X, Nie G, Zhang X. Genetic diversity and population structure analysis in a large collection of white clover (Trifolium repens L.) germplasm worldwide. PeerJ, 2021, 9(e11325.
[16]Jiang W Z, Yao F J, Lu L X, Fang M, Wang P, Zhang Y M, Meng J J, Lu J, Ma X X, He Q, Shao K S. Genetic linkage map construction and quantitative trait loci mapping of agronomic traits in Gloeostereum incarnatum. Journal of microbiology (Seoul, Korea), 2021, 59(1): 41-50.
[17]Sun M, Dong Z, Yang J, Wu W, Zhang C, Zhang J, Zhao J, Xiong Y, Jia S, Ma X. Transcriptomic resources for prairie grass (Bromus catharticus): expressed transcripts, tissue-specific genes, and identification and validation of EST-SSR markers. BMC plant biology, 2021, 21(1): 264.
[18]徐照龙, 易金鑫, 余桂红, 张大勇, 何晓兰, 王秀娥, 马鸿翔. 藜科6种耐盐植物遗传多样性的EST-SSR分析. 植物遗传资源学报. 2011, 12(01): 113-120.
[19]张金渝,杨维泽,崔秀明,金航,虞泓,陈中坚,沈涛,杨涛.三七栽培居群遗传多样性的EST-SSR分析.植物遗传资源学报.2011,12(02):249-254.
[20]Zhang F, Wang C, Li M, Cui Y, Shi Y, Wu Z, Hu Z, Wang W, Xu J, Li Z. The landscape of gene-CDS-haplotype diversity in rice: Properties, population organization, footprints of domestication and breeding, and implications for genetic improvement. Molecular plant, 2021, 14(5): 787-804.
[21]张水明,陈程,陈芳芳,汪天.16个蝴蝶兰品种EST-SSR遗传多样性分析.植物遗传资源学报.2013,14(03):560-564.
[22]信朋飞,臧巩固,赵立宁,高春生,程超华.大麻SSR标记的开发及指纹图谱的构建.中国麻业科学.2014,36(004):174-182.
[23]AXW, ALD, AQC, BD Za JGE, Conservation. Genetic diversity, population structure, and evolutionary relationships within a taxonomically complex group revealed by AFLP markers: A case study on Fritillaria cirrhosa D. Don and closely related species - ScienceDirect. 2020,
[24]Stavridou E, Lagiotis G, Kalaitzidou P, Grigoriadis I, Bosmali I, Tsaliki E, Tsiotsiou S, Kalivas A, Ganopoulos I, Madesis P. Characterization of the Genetic Diversity Present in a Diverse Sesame Landrace Collection Based on Phenotypic Traits and EST-SSR Markers Coupled With an HRM Analysis. Plants (Basel, Switzerland), 2021, 10(4):
[25]Peakall R, Smouse P E. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research--an update. Bioinformatics (Oxford, England), 2012, 28(19): 2537-2539.
[26]Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Molecular biology and evolution, 2018, 35(6): 1547-1549.
[27]Pritchard J K, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics, 2000, 155(2): 945-959.
[28]Wang M L, Zhu C, Barkley N A, Chen Z, Erpelding J E, Murray S C, Tuinstra M R, Tesso T, Pederson G A, Yu J. Genetic diversity and population structure analysis of accessions in the US historic sweet sorghum collection. TAG Theoretical and applied genetics Theoretische und angewandte Genetik, 2009, 120(1): 13-23.
[29]Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular ecology, 2005, 14(8): 2611-2620.
[30]Zheng Y, Zhang Z, Wan Y, Tian J, Xie W. Development of EST-SSR Markers Linked to Flowering Candidate Genes in Elymus sibiricus L. Based on RNA Sequencing. Plants (Basel, Switzerland), 2020, 9(10):
[31]Fan M, Gao Y, Wu Z, Zhang Q. Linkage Map Development by EST-SSR Markers and QTL Analysis for Inflorescence and Leaf Traits in Chrysanthemum (Chrysanthemum morifolium Ramat.). Plants (Basel, Switzerland), 2020, 9(10):
[32]Elam E J a R O E, Systematics. Population Genetic Consequences of Small Population Size: Implications for Plant Conservation. 1993, 24(217-242.
[33]George J, Dobrowolski M P, Van Zijll De Jong E, Cogan N O, Smith K F, Forster J W. Assessment of genetic diversity in cultivars of white clover (Trifolium repens L.) detected by SSR polymorphisms. Genome, 2006, 49(8): 919-930.
[34]Smith A L, Hodkinson T R, Villellas J, Catford J A, Cserg? A M, Blomberg S P, Crone E E, Ehrlén J, Garcia M B, Laine A L, Roach D A, Salguero-Gómez R, Wardle G M, Childs D Z, Elderd B D, Finn A, Munné-Bosch S, Baudraz M E A, Bódis J, Brearley F Q, Bucharova A, Caruso C M, Duncan R P, Dwyer J M, Gooden B, Groenteman R, Hamre L N, Helm A, Kelly R, Laanisto L, Lonati M, Moore J L, Morales M, Olsen S L, Pärtel M, Petry W K, Ramula S, Rasmussen P U, Enri S R, Roeder A, Roscher C, Saastamoinen M, Tack A J M, Töpper J P, Vose G E, Wandrag E M, Wingler A, Buckley Y M. Global gene flow releases invasive plants from environmental constraints on genetic diversity. Proceedings of the National Academy of Sciences of the United States of America, 2020, 117(8): 4218-4227.
[35]Li Z, Yun L, Gao Z, Wang T, Ren X, Zhao Y. EST-SSR Primer Development and Genetic Structure Analysis of Psathyrostachys juncea Nevski. Frontiers in plant science, 2022, 13(837787).
[36]Shen G W, Li J S, Ren C Z, Hu Y G J J O T C. Analysis of genetic diversity and population structure of oat germplasms from China and Canada. 2010,
[37]Zheng X, Cheng T, Yang L, Xu J, Tang J, Xie K, Huang X, Bao Z, Zheng X, Diao Y, You Y, Hu Z. Genetic Diversity and DNA Fingerprints of Three Important Aquatic Vegetables by EST-SSR Markers. Scientific reports, 2019, 9(1): 14074.
文章摘自:边境,王晓楠,曹焜等.大麻EST-SSR遗传结构分析及指纹图谱构建[J/OL].植物遗传资源学报:1-16[2023-08-06].DOI:10.13430/j.cnki.jpgr.20230531001.
